

前面的小编给大家详细介绍了一下TCGA这个数据库,以及如何从这个数据库下载并合并表达谱数据。然后进行差异表达分析和构建ceRNA网络。
?如何合并TCGA表达谱数据
?零代码合并TCGA表达谱数据
?零代码TCGA差异表达分析
?R代码TCGA差异表达分析
?一文掌握ceRNA网络构建
最近发现,TCGA的RNAseq这些数据似乎已经更新了。这应该是2022年4月初发生的事情。让我们来看看具体的区别。我们仍然认为CHOL以这套数据为例,解释如何下载和处理新版本TCGA中的RNAseq数据。miRNA数据没有变化。
1.打开TCGA官网https://portal.gdc.cancer.gov/.输入搜索框chol,选择第一个PR(project),TCGA-CHOL
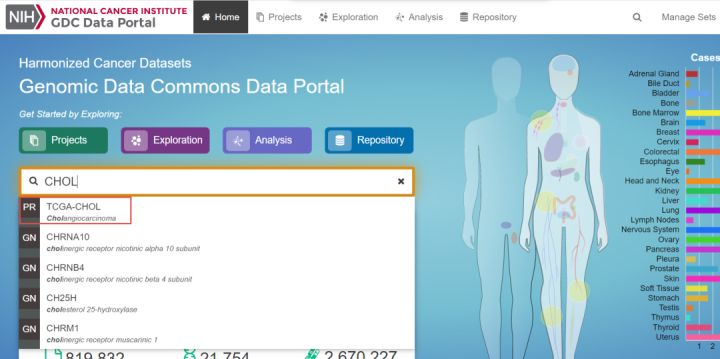
2.单击跳转页面RNA-Seq后面的数字
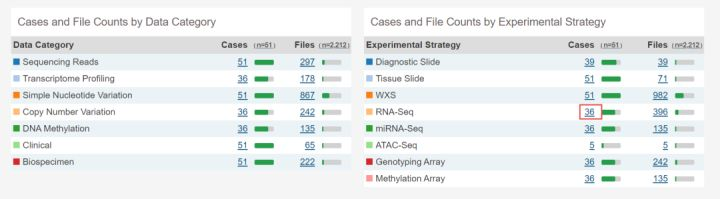
3. 点击新打开的页面左上角Files
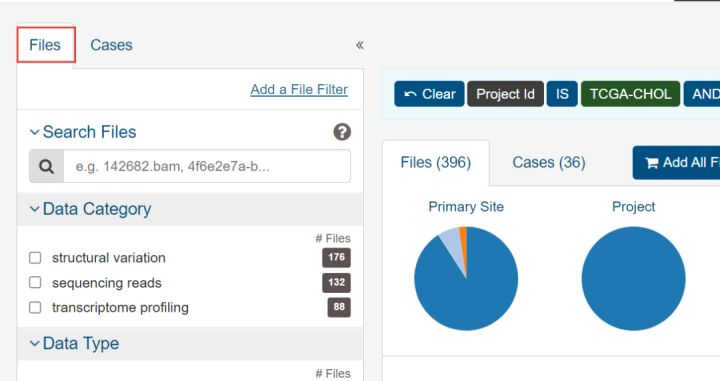
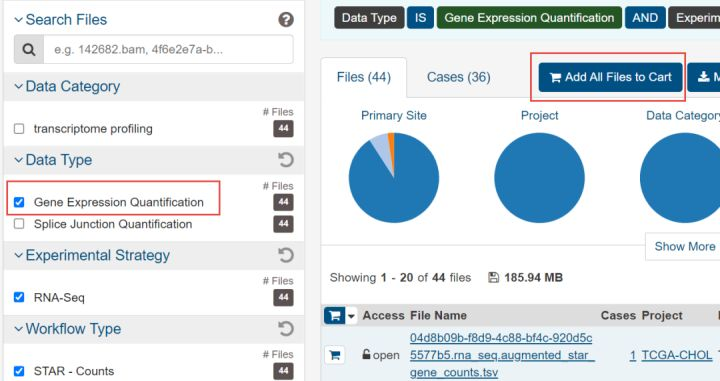
4.接下来是不同的地方,你可以看到workflow type里面没有HTSeq-Counts取而代之的是STAR-Counts。我们选择这个STAR-Counts。
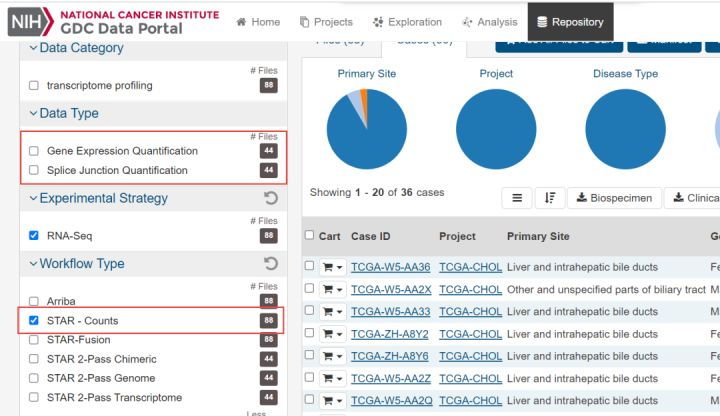
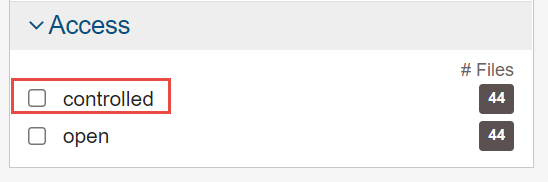
你会发现STAR-Counts有88份文件,其中44份是Gene Expression Quantification,这是我们合并表达谱所需的文件。剩下的44份文件是Splice Junction Quantification,这主要是检测新的转录本或集成文件。此外,这44份文件属于controlled下载文件需要申请权限。
5.勾选Gene Expression Quantification,点击右边的Add All Files to Cart。
6. 这个时候在我们的购物车(右上角)里面就会出现刚才选择的44个文件。
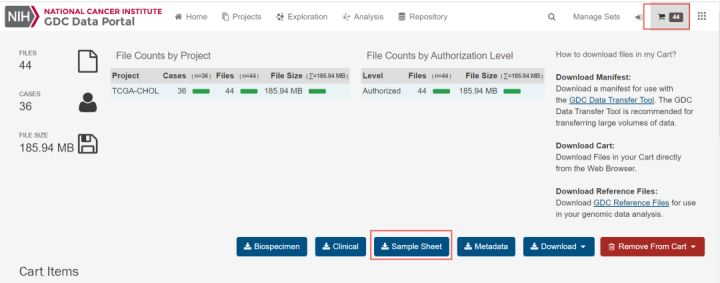
我们需要在这里下载sample sheet,点击Sample Sheet。下载的文件打开如下,可以看到新版本TCGA的counts文件的名称不再是带有的htseq.counts.gz后缀的压缩文件变成了star_gene_counts.tsv为后缀的文本文件。

还需要下载包含表达谱数据的所有内容star_gene_counts.tsv文件。点击Download,点击下拉框中的Cart。将下载压缩文件。
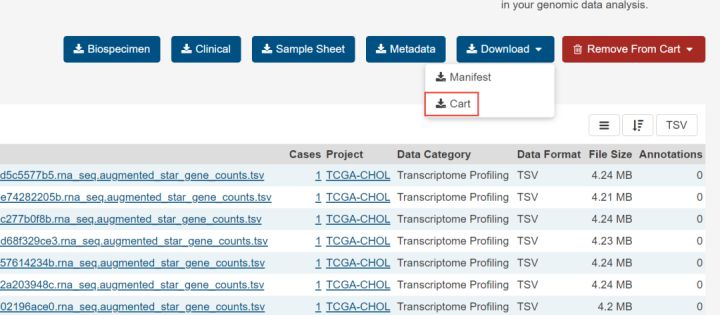
解压后会有44个文件夹

每个文件夹都有一个star_gene_counts.tsv,我们可以随意打开一个,这个文件的内容与旧版本完全不同,包含更多的信息。甚至包括RNA类型很容易区分mRNA和lncRNA此外,你不必担心基因的名称ID转换问题。
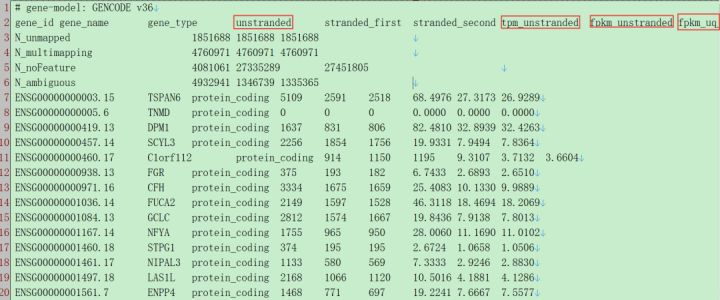
这里除了有STAR-counts,还有TPM,FPKM和FPKM_UQ。这些数据的具体计算方法可以参考TCGA官方文档https://docs.gdc.cancer.gov/Data/Bioinformatics_Pipelines/Expression_mRNA_Pipeline/
STAR-counts计算比较直接,就是有几个reads比较相应的基因,counts就是几。

TPM,FPKM和FPKM_UQ定义如下。
FPKM The fragments per kilobase of transcript per million mapped reads (FPKM) calculation aims to control for transcript length and overall sequencing quantity.Upper Quartile FPKM The upper quartile FPKM (FPKM-UQ) is a modified FPKM calculation in which the protein coding gene in the 75th percentile position is substituted for the sequencing quantity. This is thought to provide a more stable value than including the noisier genes at the extremes.TPM The transcripts per million calculation is similar to FPKM,but the difference is that all transcripts are normalized for length first. Then,instead of using the total overall read count as a normalization for size,the sum of the length-normalized transcript values are used as an indicator of size.TPM,FPKM和FPKM_UQ计算方法如下。

官方网站还给出了详细的例子,帮助您理解计算过程
Examples Sample 1: Gene AGene length: 3,000 bp1,000 reads mapped to Gene A1,000,000 reads mapped to all protein-coding regionsRead count in Sample 1 for 75th percentile gene: 2,000Number of protein coding genes on autosomes: 19,029Sum of length-normalized transcript counts: 9,000,000FPKM for Gene A = 1,000 * 10^9 / (3,000 * 50,000,000) = 6.67FPKM-UQ for Gene A = 1,000) * 10^9 / (3,000 * 2,000 * 19,029) = 8.76TPM for Gene A = (1,000 * 1000 / 3000* 1,000,000 / (9,000,000)= 37.04今天的分享先到这里,我们将介绍如何合并新版本TCGA数据库中的counts表达谱矩阵。
TCGA数据库悄悄更新—RNAseq没有HTSeq-Counts了
")
")








